The mechanism of action of the Wilms’ tumour 1 protein WT1
The Wilms’ tumour 1 protein, WT1, was first identified as a tumour suppressor in paediatric nephroblastoma. In recent years, however, it has become clear that WT1 can also act as an oncogene in several paediatric and adult cancers. Indeed, WT1 antibody therapies are showing promise in clinical trials for lung cancer, leukaemia, and other malignancies. Despite its emergence as a significant target in cancer therapy, our understanding of WT1 function at the molecular level is poor.
WT1 is a gene regulator that can activate or repress transcription, but the mechanisms by which WT1 regulates differentiation, and how it manifests tumor suppressor and oncogenic activities are not known. The long-term goal of my laboratory is to determine the molecular mechanisms that shape WT1 function in development and disease. Our studies involve the analysis of WT1 target genes and how the interaction partners of WT1 regulate its activity. Over the last few years we have identified the transcriptional repressor BASP1 and the apoptotic protease HtrA2 as major regulators of WT1 function (see figure 1). Our current studies aim to determine how BASP1 and HtrA2 regulate WT1 and thus identify new therapeutic avenues for the treatment of WT1-dependent cancers.
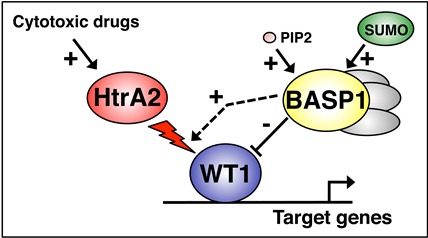
Figure 1:BASP1 (acting as part of a complex) interacts directly with WT1 at a gene promoter and converts WT1 from a transcriptional activator to a repressor. The phospholipid PIP2 is required for transcriptional repression, and this process can be augmented by sumoylation of BASP1 (SUMO). HtrA2 is an apoptotic protease that degrades WT1 upon treatment of cells with cytotoxic drugs. BASP1 can enhance this process, which is consistent with a tumour suppressor role for BASP1.