 ....
....
Synthesis and Testing of Lysine Biosynthesis Inhibitors
....
Paul Wang - Ph.D. Student 1997-2000................Rosie Stentiford M.Sci. Undergraduate Student 1997-98
Katrina Wareing - B.Sc. Undergraduate Student 1996-97
James Schouten - M.Sci Undergraduate Nuffield Summer Student 1997
Dr Russell Cox
Background
The biosynthesis of L-lysine by bacteria has been extensively studied because lysine is essential to the growth and development of these organisms, being necessary for both cell wall and protein synthesis (Fig. 1). Humans do not biosynthesise lysine, however, obtaining sufficient quantities from the diet.
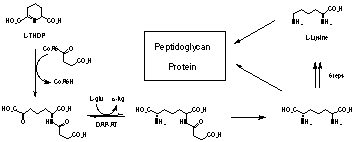
Fig. 1. Part of the bacterial L-Lysine pathway
Inhibitors of lysine biosynthesis could be good antibiotics with low human side-effects. Many of the enzymes from the bacterial lysine pathway have been investigated with the aim of developing potent and specific inhibitors. Mostly the results have been unimpressive, with inhibition constants (IC50, Ki etc.) in the mM to mM range. Recently our group has developed inhibitors with nM inhibition constants.
Summary of Recent Work by the Cox Group
Substrates of DAP-AT.
N-Succinyl diaminopimelate aminotransferase (DAP-AT, Fig. 1) is a PLP dependent enzyme from the bacterial lysine pathway. In recent work this enzyme has been isolated and purified from E. coli. We have developed a flexible synthesis of the substrate and a number of substrate analogues (Fig. 2). Measurement of kcat/Km values for these substrates has allowed an assessment of substrate specificity for the E. coli enzyme. It is clear that specificity varies with the N-acyl substituent. Some compounds such as the N-Phe-N-Ac analogue have Km values significantly lower than the natural substrate.
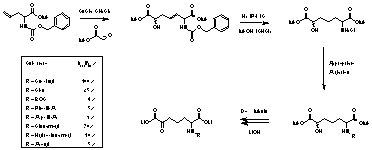
Fig. 2 Synthesis and specificity of E. coli DAP-AT substrates
Inhibitors of DAP-AT.
We reasoned that product analogues in which the product amine (NH2) was replaced by a hydrazine (NHNH2) would retain characteristics of the product to allow good enzyme binding and recognition, but as hydrazines are potent nucleophiles (and poor leaving groups) these compounds would cause inhibition (Fig. 3). These compounds were shown to be potent tight, slow-binding inhibitors of DAP-AT, with inhibition constants (Ki*) in the nM range: the best so-far reported for lysine biosynthesis inhibitors. Peptide analogues have also been synthesised and kinetic analysis has determined Ki* values. Peptide substrate analogues are also good competitive inhibitors of DAP-AT (IC50 <1mM). Antibiotic testing of the hydrazino peptides shows them to strongly inhibit the growth of E. coli in the absence of DAP and L-Lysine; the effect is attenuated on media containing these compounds.
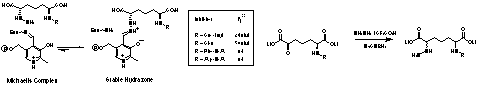
Fig. 3. Hydrazino inhibitors of DAP-AT
A second stream of inhibitors have also been designed and synthesised. Since the mechanism of PLP dependent enzymes dictates the formation of intermediate imines, compounds with leaving groups positioned adjacent to the reacting carbonyl should cause inhibition through cofactor modification (Fig. 4), or stabilisation of tetrahedral intermediates. These compounds inhibit DAP-AT as expected and kinetic analysis is currently underway to determine the mechanism and inhibition constants.
Fig 4. Oxaloyl Inhibitors of DAP-AT DAP-AT from Mycobacteria.
The purification and assay procedures adopted for E. coli DAP-AT have been applied to Mycobacterium smegmatis. A crude preparation clearly shows DAP-AT activity using the natural substrate.
Programme
Specificity of Inhibition by Hydrazino-Peptides.
A major feature of the project will be the study of the mycobacterial DAP-AT as a potential antimicrobial target. E. coli studies have already shown that the substrate N-acyl substituent is important for binding and this moiety may also be important for specificity. The project will aim to synthesise and test a range of substrate and inhibitor analogues against the mycobacterial DAP-AT in order to optimise selectivity. A combinatorial approach may be appropriate for this, especially in the formation of peptide libraries and the expertise of GlaxoWellcome would be appreciated in this respect. Our current methodology to access the hydrazines relies on the in-situ reductive hydrazination of ketones using NaCNBH3. This method allows no control of the stereochemistry of reduction and our hydrazines show little (if any) enantio-purity at the e-position. New methods of homogeneous reduction of imines and hydrazones have been developed by Burk utilising DuPhos-Rh catalysts and this methodology could be investigated for the provision of enantiomerically pure compounds. Alternatively separation of diastereomers by HPLC is a possibility.
Alternative Inhibitor Classes.
As already demonstrated, inhibitors of DAP-AT are antimicrobial compounds. An investigation of the range of inhibitor types effective against the enzyme will form a major theme of the project. In addition to the extension of the range of hydrazino-peptides proposed above, the following types of inhibitors could be synthesised and tested.
A) Oxaloyl Substrate Analogues
A small number of these compounds have already been synthesised (fig 5). These compounds are a new class of PLP dependent enzyme inhibitors. An increased range will be studied to determine whether inhibition potency varies with changes in side-chain in the same way as the hydrazino-peptides. N-oxaloyl compounds are of particular mechanistic interest as inhibition could be through stabilisation of a tetrahedral intermediate or via formation of an active site delocalised system.
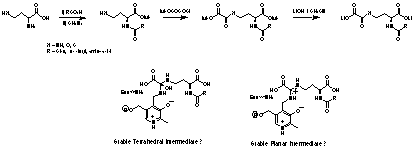
Fig. 5. Oxaloyl substrate analogue inhibitors
B) Unsaturated Substrate Analogues
b-g Unsaturated substrate analogues are usually irreversible inhibitors of PLP dependent enzymes. These compounds could be accessed via rearrangement of the b-g unsaturated ketones obtained by oxidation of the ene adduct (fig. 6). Hydrazones could also be envisaged as being irreversible inhibitors. These would be simply available by reaction of substrate analogues with hydrazine.
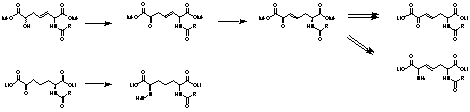
Fig. 6. Synthesis of unsaturated inhibitors
C) d-Halo Substrate Analogues
d-Halo (fluoro, chloro) substrate analogues of PLP enzymes are also usually irreversible inhibitors. These could be accessed via allylic oxidation of protected ene adducts, followed by hydroxyl displacement (fig. 7).

Fig. 7. Synthesis of d-halo inhibitors
D) Reversible Inhibitors
Our previous results have shown that reversible inhibitors (substrate analogues) are not potent inhibitors. Compounds such as a-methyl amines are not therefore likely to be of interest.
Aminotransferase Mechanism.
A later stage PLP-dependent enzyme from the Lysine pathway, DAP-decarboxylase, operates by a unique mechanism featuring unusual stereochemistry of proton transfers. This fact indicates that the enzyme may be structurally unrelated to the major classes of PLP dependent enzymes (indeed its sequence would suggest this). DAP-AT has been neither sequenced or cloned and an understanding of the stereochemical course of the reaction would aid in the design of novel substrates and inhibitors.
Inhibition Mechanism.
Inhibition by hydrazino substrate analogues is via a slow-binding mechanism with off-rate half times in the region of 45-90 min. Investigation of peptidic hydrazino-inhibitors will be carried out as the dipeptide substrate analogues show low Km values. This is suggestive of improved binding over natural substrates, indicating the possibility of lowered Ki* values for the corresponding hydrazino-inhibitors. Peptide inhibitors may also have improved membrane-transport properties which will be tested against whole cells. The oxaloyl inhibitors are a new class of PLP dependent enzyme inhibitors and their mode of action will be studied by standard kinetic methods.
Gene Cloning and Protein Expression.
In order to facilitate the above aims it will be beneficial to possess a clone of DAP-AT. The E. coli genome has recently been fully sequenced, and a search of the sequence using PLP-dependent enzyme sequences may reveal a likely candidate gene for cloning and expression. Alternatively N-terminal sequencing of purified enzyme would furnish a search sequence. A similar approach will be suitable for the mycobacterial gene as the genome sequence of M. tuberculosis is well underway.