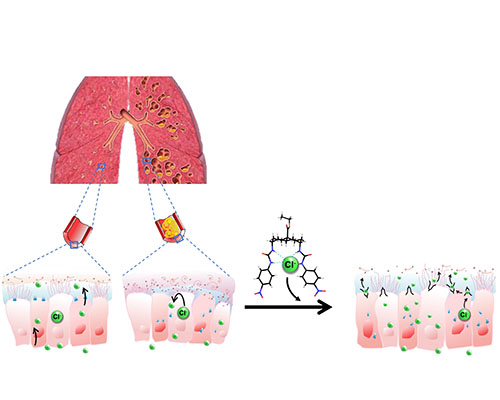
Cystic Fibrosis is a debilitating disease that leads to a build-up of sticky mucus in several organs of the body, including the lungs and intestine. It is caused by a defective gene and affects between 70,000 and 100,000 people worldwide. The disease occurs when someone possesses two faulty copies of a certain gene. This means that they lack a key molecular component at the surface of cells lining ducts and tubes in their body.
The molecule is called the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), and its job is to allow the passage of negatively charged species – anions – across the cell membrane. If CFTR is absent, the surface of cells becomes dehydrated and mucus thick and sticky, making it difficult for patients to breathe and digest food.
Most treatments, such as chest physiotherapy and antibiotics, target the symptoms to prevent or reduce complications rather than the underlying genetic defect. Some new drugs targeting faulty CFTR have recently been approved for use in England. But, these drugs are not suitable for all people with CF.
Researchers from Bristol’s Schools of Chemistry, and Physiology, Pharmacology and Neuroscience and colleagues at the University of Sydney aimed to restore transmembrane anion movement by identifying new molecules which transport anions efficaciously across cell membranes. The team designed synthetic "anion carrier" molecules to do the same job as the missing CFTR.
Using specialist equipment, the team tested the synthetic molecules’ efficacy in cystic fibrosis cells and found they were not only effective in transporting anions across the cell membranes, but also supplement the effects of new drugs targeting faulty CFTR by allowing more anion transport by cells than either the molecules or new drugs alone. These results suggest a new approach to treat cystic fibrosis.
The next stage in this research will be to test the identified molecules on sheets of cells from cystic fibrosis air passageways to determine whether they restore mucus movement and bacterial killing.
Professor Anthony Davis, Professor of Supramolecular Chemistry and one of the study’s Bristol co-authors, said: "There’s still a long way to go, but if the research proceeds as hoped it might lead to a genuinely effective and general treatment for CF patients.”
The research, which builds on previous work by the team was funded by the by the Engineering and Physical Sciences Research Council (EPSRC) and the Medical Research Council. The team would like to thank the Wolfson Bioimaging Facility team for their support.
Paper:
Anion carriers as potential treatments for cystic fibrosis: transport in cystic fibrosis cells, and additivity to channel-targeting drugsby H Li et al in Chemical Science.